
Sexual selection is a form of natural selection that favours traits that increase mating success, often at the expense of survival. It is responsible for a huge variety of characteristics and behaviours we observe in nature, and most conspicuously, sexual selection explains the elaborate ornaments such as the antlers of red deer and the tail of the male peacock. There are many theories to explain how and why these ornaments evolve; it may be a positive feedback loop of female preference and selection on males, or ornaments may signal something useful, such as the genetic quality of the male carrying them. One way or another, despite the energetic costs of growing these ornaments, and the increase risk of predation that comes with greater visibility, sexually selected ornaments must be increasing the overall fitness of individuals carrying them. They do so by ensuring the bearer gets more mates and produces more offspring.
Theory predicts that sexually selected traits should be just as likely to become larger and more ostentatious as they are to be reduced, smaller and less conspicuous. However, almost all natural examples refer to exaggerated traits. So where are all the reduced sexual traits?
Runaway Ornaments
Previous work by GEE researchers Dr Sam Tazzyman, and Professor Andrew Pomiankowski has highlighted one possible explanation for this apparent imbalance in nature – if sexually selected traits are smaller, they are harder to see. Using mathematical models, last year they showed that differences in the ‘signalling efficacy’ of reduced and exaggerated ornaments was sufficient to explain the bias we see in nature. Since the purpose of sexually selected ornaments is to signal something to females, if reduced traits tend to be worse at signalling, then it makes sense that they would rarely emerge in nature. Their model covered the case of runaway selection, whereby sexually selected traits emerge somewhat spontaneously due to an inherent preference in females. It goes like this – if, for whatever reason, females have an innate preference for a certain trait in males, then any male who randomly acquires this trait will get more mates and produce more offspring. Those offspring will include males carrying the trait and females with a preference for the trait, and over time this creates a feedback loop that can produce extremely exaggerated traits. Under this model of sexual selection, differences in the signalling efficacy can be sufficient to explain why we so rarely see reduced traits.
Handicaps
 However, this is just one model for how sexually selected ornaments can emerge, so this year GEE Researchers Dr Tazzyman and Prof Pomiankowski, along with Professor Yoh Iwasa from Kyushu University, Japan, have expanded their research to include another possible explanation – the handicap hypothesis. According to the handicap principle, far from being paradoxical, sexually selected ornaments may be favoured exactly because they are harmful to the individual who carries them. In this way, only the very best quality males can cope with the costs of carrying huge antlers or brightly coloured feathers, and so the ornament acts as a signal to females indicating which males carry the best genes. This is an example of honest signalling – the ornament and the condition or quality of the carrier are inextricably linked, and there is no room for poor quality males to cheat the system.
However, this is just one model for how sexually selected ornaments can emerge, so this year GEE Researchers Dr Tazzyman and Prof Pomiankowski, along with Professor Yoh Iwasa from Kyushu University, Japan, have expanded their research to include another possible explanation – the handicap hypothesis. According to the handicap principle, far from being paradoxical, sexually selected ornaments may be favoured exactly because they are harmful to the individual who carries them. In this way, only the very best quality males can cope with the costs of carrying huge antlers or brightly coloured feathers, and so the ornament acts as a signal to females indicating which males carry the best genes. This is an example of honest signalling – the ornament and the condition or quality of the carrier are inextricably linked, and there is no room for poor quality males to cheat the system.
Using mathematical models, the authors investigated four possible causes for the absence of reduced sexual ornaments in the animal kingdom. Firstly, like the case of runaway selection, differences in signalling efficacy might explain the bias. Under the handicap hypothesis, ornaments act as signals of genetic quality, so it would be little surprise that their visibility or effectiveness at conveying the signal would be important. Smaller ornaments may simply be worse at attracting the attention of females, meaning that the benefits of the sexual ornament don’t outweigh the costs. Similarly, the costs for females of preferring males with reduced ornaments may be higher than for exaggerated ornaments, because it is easier to find males with exaggerated traits. Again, this could theoretically tip the balance of cost and effect away from selecting for reduced ornaments. An alternative explanation is that the costs of the ornament itself are different for reduced and exaggerated traits. This seems quite likely in many cases, since a large ornament would require more resources to grow. But in this case selection would be more likely to produce reduced ornaments with lower costs! In order to replicate the excess of exaggerated traits we see in nature, reduced traits would have to cost more – much less biologically plausible. However, if large ornaments tend to be more costly, then they may be more likely to be condition-dependent, a key tenant of the handicap principle. Exaggeration may be more effective at producing honest signalling and exaggerated traits may therefore be more useful to females as a signal of quality.
Honest Signals
The results of modelling highlighted two key ways in which exaggerated traits might be favoured by the handicap process. In the model, when exaggerated traits have a higher signalling efficacy or are more strongly condition-dependent, exaggerated traits tend to be more extreme than reduced traits. The model still predicts that reduced traits are equally likely to evolve, just that they will tend to be less extreme examples of ornamentation. The other two possible explanations – higher costs to females that prefer small ornaments or the males that carry them, failed to consistently produce the observed lack of reduced ornaments. Both explanations were able to produce this outcome under certain circumstances, but in other circumstances they produced the opposite effect. Exaggerated ornaments, therefore, may be more common because they are more effective signals that are more likely to be honest.
Based on this and previous work by Dr Tazzyman and colleagues, asymmetries in the signalling efficacy of reduced and exaggerated traits is sufficient to explain the lack of reduced traits in nature. Whether ornaments evolve via runaway selection or the handicap process, asymmetrical signalling efficacy tends to favour exaggerated traits. However, in the case of the handicap process, asymmetries in condition-dependance of the trait may also be involved. These two explanations are not mutually exclusive, and it is likely that in reality many factors are involved.

Importantly, for both explanations and for both type of sexual selection, the models still predict that exaggeration and reduction will be equally likely. The differences emerge in terms of how extreme the ornament will become. Thus, this work predicts that there are many examples of reduced ornaments in nature, perhaps we just haven’t found them yet. This is especially likely if reduced traits that might be less noticeable anyway also tend to be less extreme. Alternatively, there may be other asymmetries not yet considered that make reduced ornaments less likely to emerge in the first place.
Where Next?
The authors suggest some very interesting avenues for future research. Firstly, they suggest that more complex models investigating how multiple different asymmetries may act together to produce sexually selected ornaments will get us closer to understanding the intricate dynamics of sexual ornamentation. Secondly, these models have only considered cases where evolution of the trait eventually settles down – at a certain ornament size, the costs and benefits of possessing it are equal, and the trait should remain at this size. However, in some cases the dynamics are more complex, and traits undergo cycles of exaggeration and reduction. Research into the impact of asymmetries in condition-dependence and signalling efficacy in these ‘nonequilibrium’ models would yield fascinating insights into the evolution of sexual ornaments.
Original Article:



This research was made possible by funding from the Natural Environment Research Council (NERC), the Engineering and Physical Sciences Research Council (EPSRC), the European Research Council (ERC)
 Close
Close

 Sloths might be notorious for their leisurely pace of life, but research published last year shows they are no slow coaches when it comes to evolution.
Sloths might be notorious for their leisurely pace of life, but research published last year shows they are no slow coaches when it comes to evolution.

















 So, UPI makes a lot of sense, evolutionarily, and some scientists think it might also explain why we have two sexes, as opposed to any other mating system. It’s important to be clear, when we talk about having two sexes we’re saying nothing about the external differences between the sexes (sexual dimorphism) observed in many multicellular organisms. We’re talking about the existence of two ‘mating types’, such that individuals cannot mate with members of the same type. Recent research by another group of GEE academics including Professor Andrew Pomiankowski, Dr Nick Lane and Professor Robert Seymour, investigated the evolution of UPI and in particular it’s relationship with the evolution of a two-sex mating system. We might expect a strong link between UPI and the existence of two sexes, since uniparental inheritance immediately generates differences between the two mating partners, and ensures that reproduction is not possible unless one member of each ‘type’ is present. Although UPI is often thought to have been a key driver in the evolution of mating types, there have been few investigations of what conditions are needed for the fitness benefits of UPI to actively drive the emergence of two mating types. So the authors developed a new mathematical model of mitochondrial inheritance and the evolution of UPI in a population where biparental inheritance (BPI) is the norm. They incorporated mitochondrial mutation (which might sometimes be selfish) and selection into the model, and included different mating types.
So, UPI makes a lot of sense, evolutionarily, and some scientists think it might also explain why we have two sexes, as opposed to any other mating system. It’s important to be clear, when we talk about having two sexes we’re saying nothing about the external differences between the sexes (sexual dimorphism) observed in many multicellular organisms. We’re talking about the existence of two ‘mating types’, such that individuals cannot mate with members of the same type. Recent research by another group of GEE academics including Professor Andrew Pomiankowski, Dr Nick Lane and Professor Robert Seymour, investigated the evolution of UPI and in particular it’s relationship with the evolution of a two-sex mating system. We might expect a strong link between UPI and the existence of two sexes, since uniparental inheritance immediately generates differences between the two mating partners, and ensures that reproduction is not possible unless one member of each ‘type’ is present. Although UPI is often thought to have been a key driver in the evolution of mating types, there have been few investigations of what conditions are needed for the fitness benefits of UPI to actively drive the emergence of two mating types. So the authors developed a new mathematical model of mitochondrial inheritance and the evolution of UPI in a population where biparental inheritance (BPI) is the norm. They incorporated mitochondrial mutation (which might sometimes be selfish) and selection into the model, and included different mating types. 


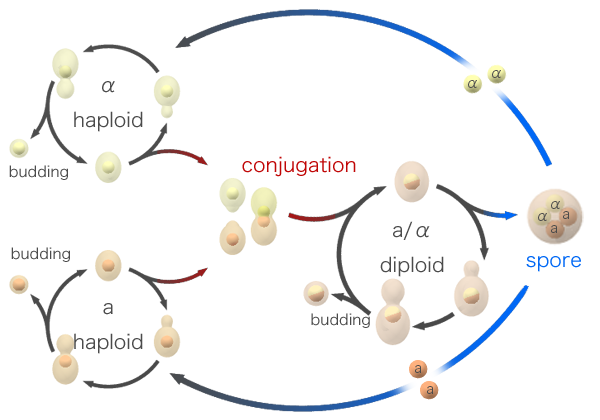 Yeast, being single-celled, are able to reproduce asexually – budding off a new copy of themselves. However, when times are hard, yeast can also enter a sexual phase of reproduction. When nutrient levels are low, a yeast cell will divide into sexual spores. These spores are haploid, carrying only one copy of each chromosome (rather than the usual two), making them analogous to sperm and egg cells. Spores will remain dormant until they sense an improvement in the environment, at which point they spring into action and try to find a mate. Spores come in two mating types and must always mate with another spore of the opposite type – a system based on mating pheromones released by spores to attract the opposite sex.
Yeast, being single-celled, are able to reproduce asexually – budding off a new copy of themselves. However, when times are hard, yeast can also enter a sexual phase of reproduction. When nutrient levels are low, a yeast cell will divide into sexual spores. These spores are haploid, carrying only one copy of each chromosome (rather than the usual two), making them analogous to sperm and egg cells. Spores will remain dormant until they sense an improvement in the environment, at which point they spring into action and try to find a mate. Spores come in two mating types and must always mate with another spore of the opposite type – a system based on mating pheromones released by spores to attract the opposite sex.

