 Close
Close

Beyond the data revolution
By regfbec, on 31 December 2017
The data explosion has become a media cliché, affecting almost all aspects of contemporary life. In the biomedical sphere, the “omics” revolutions are continuing to generate terabytes of biomolecular data, increasingly supplemented by multiple other enormous data sets, often linked to each other, which include images, and a growing set of behavioural and clinical parameters. It is a truism that computational tools are required to process and interpret this data, and as I explored in a previous blog, training individuals who can work at the interface between computer science and biomedical science is one of the major challenges in medical and scientific training. But two important problems still limit the impact of the data revolution on the progress of biomedical research and its eventual translation into improved health.
The first recurring problem, common to research centres around the world, is how best to integrate the individuals with the computational skills into the biomedical research enterprise. Computer scientists and life scientists are trained to speak in a different language, and possess quite different skill sets. It is a long and arduous process to train a mathematician or computer scientist to truly understand the nature of the problem being worked on by their biologist colleagues. And it is at least as difficult, and arguably even harder, for a life scientist to take the reverse path. In many institutions, individuals providing computational skills act as data analysts, providing a service to one or more research groups. Indeed this has led to the emergence of new specialised bioinformatics training courses. Such core facilities have proved invaluable, especially to provide low level data processing, and acting as the human interface between the data generating machine and the biologist wanting the data. Nevertheless, this model does not harness the full power of computational science to the biomedical enterprise. The interests and excitement which drives the scientists who are creating the extraordinary current explosion in computer science are usually not captured by providing a largely technical service to their life science colleagues. And individuals in such centres are often stretched, working on many simultaneous projects, and with little time and energy to integrate cutting edge computer science research into their everyday analysis.
The second problem exists at the level of the data itself. From one point-of-view, the high dimensional complex data sets being generated today can be regarded simply as extensions of the simpler oligo-molecular studies of classical biology. RNAseq, for example, certainly allows one to capture the levels of thousands of transcripts, rather than one or two at a time. But after the raw data has been converted into gene lists, analysis often involves homing in on selected genes or sets of genes of ‘interest’ and basically ignoring the rest of the data. Similar considerations apply to many other examples of ‘big data’. But without a clear conceptual framework for handling the data as a cohesive data sets, and not as a series of measurements on many individual and unrelated parameters, it is hardly surprising that the laboratory scientist is overwhelmed by the overwhelming flow of new data. All too often, the computational scientist working on a project is challenged with the question ‘what does this data mean?”. The biologist almost expects some magical interpretation which will reveal some fundamental but unknown answer to an unasked question lying buried within the data set. But computer scientists are not magicians !
I would like to propose that mathematical modelling can offer some of the answers to both the challenges outlined in the previous two paragraphs. In applied mathematics, statistics, computer sciences and indeed the physical sciences in general, model building is at the very heart of the scientific enterprise. Such models are of many types : statistical models, mechanistic models of physical processes, deterministic or stochastic models, agent-based models etc. They can be used to link theory and application. The complexity of model output often outstrips the intrinsic complexity of the model itself. Even an ordinary differential equation, by the addition of a simple time delay term, can produce output of ‘chaotic’ complexity. Nevertheless, models provide ways of describing, exploring and capturing the behaviour of real life processes in a way which no other approach can do. And they provide a focus for linking theory and application, a nexus for the combined efforts of biologist, mathematician and computer scientist.
The introduction of modelling into the life sciences is, of course, not new. Enzymology, pharmacology, genetics, structural biology : there is a long list of subjects in which models have played fundamental roles. But today we need to work on new classes of models which capture and deal with big data. Systems biology has begun to provide such models. Of course, at one level a model provides testable hypotheses and suggest new experiments which can be used to verify or falsify the model. This cycle between model building and experimentation is well accepted. But at a much deeper and more general level, models can provide the essential link between data and biological understanding : they provide a way to understand large data sets at the scale of the data itself. They provide an antidote to reductionist molecular biology, which has provided the spectacular advances of the past century, but which is struggling in the face of the current data flood. At the same time, it can provide the focal point for drawing together the physical and life scientists. Mathematicians and computer scientists alike will be excited and drawn in by the models which speak in a language they can understand, and provide challenges they are fully equipped to explore. Indeed, exploring models which capture and encompass ‘big data’ is one of the major challenges in contemporary computer science. But a good model will also inspire and motivate the experimental scientist. Models can provide the common talking point which will drive true integration of mathematics, computer science and biology.
PS These thoughts arose as I was preparing a new teaching module called “Mathematical modelling in biomedicine” which will start in January 2018.
Systems biology : making sense of complexity
By regfbec, on 14 December 2017
Register now at https://systemsbiologysymposium.eventbrite.co.uk
 Loading...
Loading...
Mathematics, computers and medicine
By regfbec, on 6 February 2017
The eminent 19th Century physician Sir William Osler, the founder of the John Hopkins School of Medicine in Baltimore was famously quoted as having said “Medicine will become a science when doctors learn to count”. Sadly, his prophetic words have had little impact. Mathematics, and its younger partner, computer science make barely any contribution to the current medical school curriculum in most universities, just as they did not 150 years ago. And yet medicine is now in the midst of a revolution which may be as profound as the impact of molecular biology in the second half of the last Century. A revolution driven primarily by the extraordinary developments in computers and processing power. Scarcely a field of medicine remains untouched. Primary care provision is coming to terms with vast new data sets comprising comprehensive electronic patient records which will link an individual’s genetic makeup to behavioural and social data and medical history. Miniaturisation and price reduction are boosting point-of-care diagnostics and real-time physiological monitoring. A score of new imaging modalities which leverage the latest advances in artificial intelligence and machine learning are driving the emergence of computational anatomy, providing real time dynamic images of every part of the human body. Robotics are transforming surgery. Genomics, proteomics and metabolomics are creating precision medicine. The list is endless.
Computers and mathematical models lie at the heart of all these advances. Computers are needed to drive the new instrumentation, and then to capture, store and organise the vast data sets they generate. But generating, storing and classifying data are not enough. The real benefits can only arise from the ability to extract the underlying biology and pathology which are embedded in this data. This ability, in turn, requires mathematical modelling : the ability to recognise and formulate the rules and relationships which allow the data to make sense, which enable us to predict the future based on the past, and which ultimately will guide and inform the clinician and establish best medical practice.
The mathematics may be of many different types. It may be classical mathematics, fields such as classical , statistical or quantum mechanics, linear and non-linear differential equations, probability theory, the mathematics of light etc. Or it may be newer branches of mathematics, fields which some mathematicians may not even recognise as legitimate branches of mathematics at all: artificial intelligence, machine learning, high dimensional statistics, computational algorithms etc. Indeed so fluid and dynamic is the interface between mathematics, computer science and biology and medicine that it would be a brave individual who would predict with any confidence the pace or direction of progress which we may observe in the coming decades.
So how can we prepare the next generation of doctors for this brave new world, where they will properly be able to exploit the data and computational revolutions which are already transforming medical care in the 21st Century ? Clearly mathematics and computation will gradually need to be embedded in the medical school curriculum, in the same way that molecular biology and biochemistry have now taken their rightful place alongside the classical study of anatomy and pathology. UCL, one of the top medical schools in the UK, with a long-established commitment to train doctors who are at the cutting edge of medical advances, has recently taken an important step in this direction. UCL’s Programme and Module Approval Panel has recently approved my proposal for a new Intercalated BSc degree in Mathematics, Computers and Medicine, which will be launched in the academic year 2018/2019. The one year degree will be offered as an option in the third year of the medical school curriculum, when all students at UCL have to chose a BSC in a subject of their choice. The course will be interdisciplinary, and will be run jointly by the Faculties of Medical Sciences and the Faculty of Engineering. It will provide teaching in advanced mathematics (capitalising on the fact that a substantial portion of the entry into medical school have an A in Mathematics at A level) , computer programing , mathematical modelling and analysis of big data. Perhaps of greatest importance, the new degree will capitalise on the wealth of interdisciplinary research which already occurs at UCL to offer student research projects at the frontier between biomedicine, computers science and mathematics.
The new degree is a small step in the necessary reform of the medical school curriculum to keep pace with the computer revolution. The graduates will not necessarily become adept mathematicians or computer scientists, just as most of the graduates of the other IBScs do not spend their careers as laboratory scientists. But it will provide a core group of new talented doctors, who have confidence in their ability to understand and interact with mathematicians and computer scientists, and who will provide the leaders in the computational revolution of medical practice.
Two new jobs at the Chain lab. !! Please forward to anyone who might be interested,
By regfbec, on 8 January 2017
Position 1 http://www.jobs.ac.uk/job/AWG425/research-technician/
Research Technician
University College London – Cancer Institute / Research Department of Cancer Biology
Location: London
Salary: £34,056 to £41,163 per annum, inclusive of London Allowance, UCL Grade 7
Hours: Full Time
Contract Type: Contract / Temporary
Placed on: 19th December 2016
Closes: 15th January 2017
Job Ref: 1619025
A position for Research Technician is available to carry out RNAseq and T cell receptor sequencing on clinical samples for a variety of projects related to tumour immunotherapy.
The postholder will work alongside existing research technicians to help run the Pathogen Genomics Unit (PGU), a unique facility providing pathogen genomics, transcriptional profiling and T/B cell receptor repertoire sequencing. The role will involve processing samples for next generation sequencing and adapting existing library preparation pipelines for high throughput robotics. The postholder will receive training in all aspects of sample processing and next generation sequencing.
The post is funded by the Cancer Research UK Cancer Immunotherapy Accelerator (CITA) Network for twenty four months in the first instance.
Position 2 http://www.jobs.ac.uk/job/AWM627/research-associate/
Research Associate
University College London – UCL Division of Infection and Immunity
Location: London
Salary: £34,056 to £41,163 per annum (UCL Grade 7), inclusive of London Allowance.
Hours: Full Time
Contract Type: Contract / Temporary
Placed on: 6th January 2017
Closes: 24th January 2017
Job Ref: 1620086
Applications are invited for a Postdoctoral Research Associate in the Division of Infection & Immunity at UCL, funded by Unilever PLC, and available immediately, to work on an analysis of the T cell receptor repertoire in the context of allergic contact dermatitis.
The postholder will be supervised by Professor Benny Chain and be based in Cruciform Building. Prof Chain works closely with members of the Computer Sciences Department at UCL, to develop algorithms with which to study high dimensional immunological data sets, with a special emphasis on T cell receptor repertoire (see https://blogs.ucl.ac.uk/innate2adaptive/ and www.innate2adaptive.com) The postholder will also have the opportunity to interact with other groups within the Unilever led consortium, including in-house modeling expertise within the toxicology group at Unilever.
The post is available until 31st December 2017 in the first instance. Further funding may then become available.
How quantitative is quantitative immunology ?
By regfbec, on 9 March 2016
I recently attended a workshop on Quantitative Immunology held under the auspices of the Kavli Institute of Theoretical Physics at the University of California, Santa Barbara (UCSB). The 30 or so participants came for a variety of backgrounds : the majority from physics, a fair number of classical experimental immunologists and a handful of applied mathematicians. The meeting, which lasted three weeks, was structured round seminars given by the participants, but these provided the focus for lots of interaction, discussion and debate. In refreshing contrast to most conventional immunology conferences, the emphasis was very much on ideas rather than overwhelming displays of data. I would like to consider briefly what themes emerged from the meeting, and how these may help define the scientific space of Quantitative Immunology.
Evolution, in many guises, was a nexus joining the realms of physics and biology. Mutation/selection, in the evolutionary sense was invoked at a molecular level to shape antibody and T cell repertoires, at a cellular level to create the naïve and memory compartments of adaptive immunity populations, and at a systems level to form entire signalling networks. The ability to apply well developed stochastic models proved an enticing approach for physicists to enter the world of immunology. Quantities borrowed directly from physics (information content, entropy) , or from ecology (e.g. diversity) were frequently used although the meaning of these terms in the immunological context would have benefited from more precise explanation.
The meeting was full of interesting approaches and I certainly learnt a lot about how physicists might approach the complicated world of classical immunology. But I was struck by how often the emphasis on the “quantitative” was absent. There were of course exceptions. The careful measurements of IL2 consumption/diffusion Oleg Krichevsky; the probabilistic models of TCR generation (Thierry Mora and Aleksandra Walczak) , the kinetic proof reading models of TCR transduction (Paul Francois) , and the simple but elegant birth/death models of repertoire generation (Rob de Boer) were examples (just a few of my favourites and not meant to be an exclusive list!) of modelling which generated precise quantitative predictions amenable to experimental measurement and verification/falsification.
But often, quantitative model parametrisation seemed almost an afterthought, and quantitative prediction not really the goal. Instead, models were used as conceptual tools to describe and hence gain intuition into very complex biological systems. This type of modelling can surely be helpful. But it is a long way from the precise equations of physics, based on a handful of fundamental “constants” and whose power lies precisely in their ability to precisely predict quantitative behaviour of a physical system to the limits of measurable accuracy available.
The impression I came away with is that bringing immunology into the quantitative era is still a project in its infancy. Of course, data of a more-or-less quantitative nature is being produced at an exponentially increasing rate : single cell data, flow cytometric data, genomics data, transcriptomics data. But we lack a conceptual framework which will somehow capture this data in mathematical formulae, which can be predictive, accurate and offer intuitively appealing interpretations of immunological complexity.
What causes HIV-induced immunodeficiency?
By regfbec, on 11 January 2016
Chronic HIV infection almost always leads to a progressive immunodeficiency. The manifestations of the immunodeficiency are quite characteristic to HIV infection. Specifically, commensal or non-pathogenic environmental micro-organisms, which are generally contained by the immune system asymptomatically, lead to uncontrolled infections, morbidity and, if untreated, death. Interestingly, although HIV infection leads to a specific loss of CD4 cells, many of the major HIV-associated infections are caused by intracellular organism, both viral (e.g. KSHV, HSV, CMV), bacterial (e.g. Mycobacterium) and fungal (e.g. Cryptococcus), infections in which the CD8 T cell response has an important protective role. The mechanisms which underlie HIV pathogenesis continue to be hotly debated. Functional defects have been reported not just in the primary target of HIV (CD4 T cells) but in almost every other facet of immunity. But unravelling which changes cause immunodeficiency, and which are the indirect consequence of it remains an unsolved challenge.
We have recently re-examined the T cell repertoire in chronic untreated HIV infection using high throughput sequencing to generate a global picture of alpha and beta gene usage (Frontiers in Immunology). We observe multiple levels of immune dysregulation, including a decrease in overall diversity, which seems to be driven both by a reduced CD4 cell repertoire, but also by a very extensive over expansion of individual CD8-associated T cell clones which take up a progressively larger proportion of the available repertoire space. We repeated our analysis on samples taken from the same patients three-four months after they started ART treatment, by which time viral load was very low or undetectable in all patients. Clinical experience has established that this short treatment period leads to dramatic improvements in patients suffering from HIV-associated infections at the time of treatment initiation, suggesting that even this brief time is sufficient for significant functional repair of immune function. As expected, CD4 T cell numbers were still very low at the time of the second sample, and we observed only a very small increase in overall repertoire diversity. However, the ability to quantitatively track the frequency of individual alpha and beta genes in the two sequential samples, showed a remarkably dynamic picture underlying the apparent stable overall T cell numbers and diversity (see fig 5 in paper). Many of the TCRs which were highly expanded in the HIV+ samples showed decreases in abundance of 100 fold or more, while other TCRs increase in abundance. These dramatic changes in T cell receptor abundance are much larger than are observed when comparing paired samples taken at similar time intervals from healthy volunteers. Although the repertoire method we used does not allow pairing between alpha and beta chains, we were able to identify a number of TCR genes present in the HIV+ samples which had been characterised as forming part of HIV specific TCRs in previous studies. These sequences generally showed a fall in abundance following three months of therapy, in agreement with the rapid decrease in HIV specific T cells previously observed to accompany the ART induced fall in viral load.
On the basis of these observations, we propose a model linking dysregulation of TCR repertoire to immunodeficiency. The model remains speculative, but we hope it will stimulate further discussion and experimental verification. We propose that HIV infection drives an unregulated expansion of specific CD8 T cells, responding either directly to chronic exposure to HIV antigens, or to gut microbial antigens which enter the circulation as a result of HIV induced damage to the gastrointenstinal barrier . The overexpansion of these clones distorts the CD8 T cell repertoire, and may specifically out-compete some of the CD8 T cells responsible for containing the growth of microorganisms to which we are commonly exposed or by which we are already chronically but asymptomatically infected. As a result, organisms which are normally harmless can no longer be adequately controlled and lead to overt infection and morbidity. The rapid fall in viral load following ART leads to a rapid decrease in the abundance of these over-expanded clones (as antigen load diminishes). This in turn leads to a normalisation of the CD8 effector repertoire, and hence reversal of functional immunodeficiency and control of facultative pathogens long before the slow regeneration of the CD4 compartment can take place.
Heterogeneity in the PCR
By regfbec, on 13 October 2015
Unexpected heterogeneity in the PCR highlights the importance of molecular barcoding in immune repertoire studies. Benny Chain and Katharine Best Read the paper here
The polymerase chain reaction (PCR) is probably the most widely used technique in biology today. It is based on the process which defines all living organisms, namely the template driven replication of a nucleic acid chain by a polymerase enzyme. Since each piece of DNA gives rise to two daughter strands at each cycle, the amplification is exponential. This gives PCR extraordinary sensitivity (a single DNA molecule can readily be detected). However, it also means that the degree of amplification achieved is very sensitive to the efficiency of amplification, namely the proportion of strands which are correctly replicated at each step. This efficiency is well known to depend on a whole variety of experimental factors, including the sequences of both oligonucleotide primers and template DNA. Traditional approaches to quantitative PCR therefore relied on measuring the average amplification of an unknown number of molecules of a specific target sequence, in comparison to known numbers of a reference sequence. However, the advent of massively parallel sequence technologies now allows one to actually count the number of times each a particular sequence is present in a PCR amplified mixture. Indeed counting heterogeneous DNA mixtures by sequencing lies at the heart of RNA-seq transcriptional profiling, and especially at attempts to describe the adaptive immune repertoire by T cell or B cell receptor sequencing. We have therefore re-examined the heterogeneity of PCR amplification using a combination of experimental and computational analysis.
In our paper we label each template molecule of DNA with a u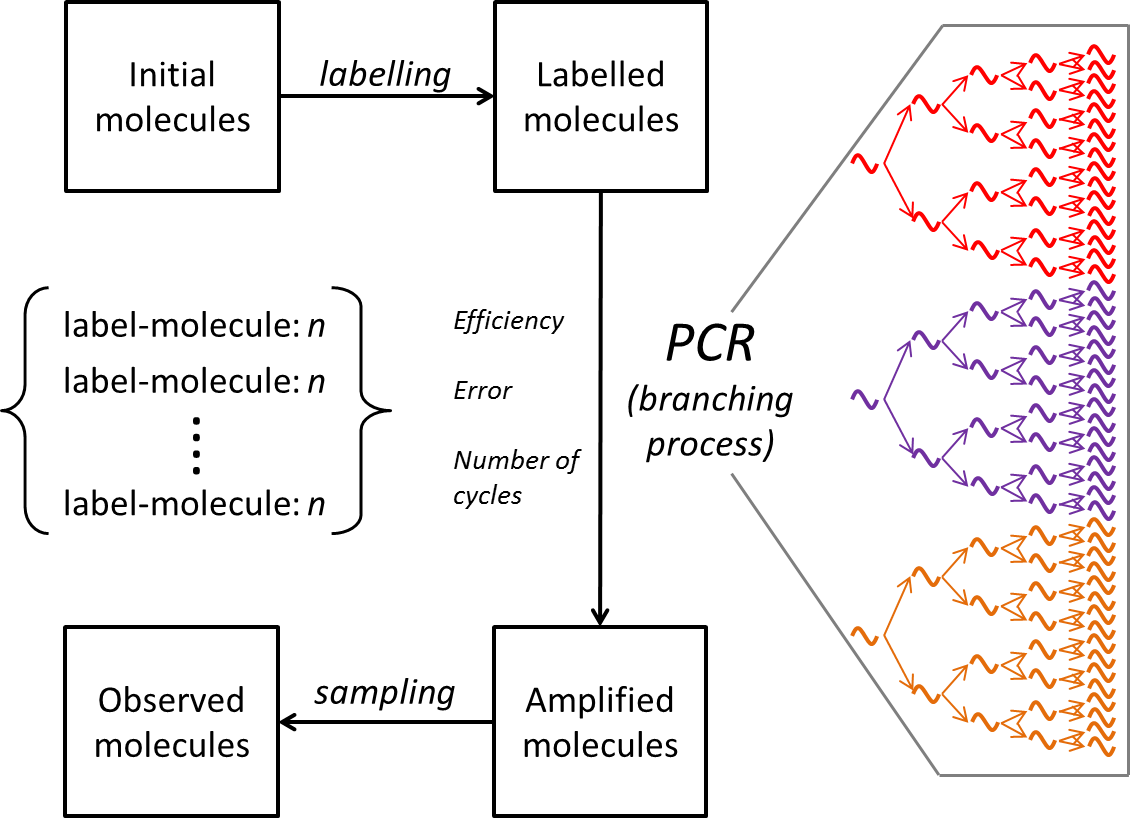 nique sequence identifier (a molecular barcode), amplify by PCR and count the number of times each DNA molecule is replicated using Illumina parallel sequencing. Remarkably, identical molecules of DNA, amplified within the same reaction tube, can give rise to numbers of progeny which can differ by orders of magnitude. PCR is an example of a branching process (see adjacent figure ). The mathematical properties of such processes have been studied quite extensively, but turn out to be complex. Indeed equations describing the processes cannot be obtained in closed form except in certain very limited specific cases. Instead, we build a “PCR” simulator (the code for which is freely available) which carries out PCR reactions in silico, tracking the fate of each molecule though a series of cycles. Using the simulator, we demonstrate that the observed heterogeneity can only be explained if different molecules of identical DNA are replicated with different efficiencies, and these differences are inherited by the progeny at ach cycle. The physical explanation of this heterogeneity remains mysterious. But these results highlight that the results of single molecule counting following PCR must be treated with caution, and highlight the importance of molecular barcoding in achieving reliable quantitative descriptions of complex molecular mixtures such as those representing the vertebrate adaptive immune repertoire.
nique sequence identifier (a molecular barcode), amplify by PCR and count the number of times each DNA molecule is replicated using Illumina parallel sequencing. Remarkably, identical molecules of DNA, amplified within the same reaction tube, can give rise to numbers of progeny which can differ by orders of magnitude. PCR is an example of a branching process (see adjacent figure ). The mathematical properties of such processes have been studied quite extensively, but turn out to be complex. Indeed equations describing the processes cannot be obtained in closed form except in certain very limited specific cases. Instead, we build a “PCR” simulator (the code for which is freely available) which carries out PCR reactions in silico, tracking the fate of each molecule though a series of cycles. Using the simulator, we demonstrate that the observed heterogeneity can only be explained if different molecules of identical DNA are replicated with different efficiencies, and these differences are inherited by the progeny at ach cycle. The physical explanation of this heterogeneity remains mysterious. But these results highlight that the results of single molecule counting following PCR must be treated with caution, and highlight the importance of molecular barcoding in achieving reliable quantitative descriptions of complex molecular mixtures such as those representing the vertebrate adaptive immune repertoire.
Benny Chain and Katharine Best
Are anti-self responses ubiquitous and necessary for a successful immune response ?
By regfbec, on 7 August 2015
Some of you might be interested in a new paper which we have just published in Frontiers in Immunology (link). The paper outlines a new model built on cellular cooperation to explain the establishment of peripheral T cell tolerance. Unusually, the paper is wholly theoretical. The paper is also somewhat technical, since the model is formulated mathematically as a linear programing optimization problem that can be implemented as a multiplicative update algorithm, which shows a rapid convergence to a stable state. We (i.e. the authors) would welcome feedback on the paper, as it represents the first step in a more ambitious plan to build a broader theoretical framework underpinning the human immune system.
The topic I want to discuss here, however, relates to some fundamental predictions of the model, with far reaching implications across many areas of immunology. In brief the model predicts that anti-self T cells remain present as part of the normal self-tolerant T cell repertoire. The recognition that cells with the potential to react against self are present in normal healthy people (i.e. thymic deletion is not complete) is now widely accepted. A particularly striking example is the latest paper from Mark Davis’s lab. http://www.ncbi.nlm.nih.gov/pubmed/25992863 ). The model further predicts, however, that some of these anti-self T cells will become activated (to a greater or lesser extent), whenever an immune response to a foreign antigen (e.g. a pathogen) is initiated. I formulate the possible significance of these predictions as three clear and testable hypotheses :
- T cells carrying receptors which recognise and respond to self are activated whenever there is a response to a foreign antigen.
- The dynamics of anti-self and anti-foreign T cell activation and proliferation will generally be different, such that tolerance to self is usually rapidly re-established.
- The self-responsive T cells play a key role in amplifying and strengthening normal responses to foreign antigen.
If correct, these hypotheses have wide-ranging implications for many aspects of immunity. I select just three settings in which anti-self responses may play a key role in immune protection or immune pathogenesis:
- Cancer. The extraordinary successes of anti-checkpoint inhibitors in many types of cancer suggest a paradox for conventional theories of specific antigen-driven immunity. There is increasing evidence that the efficacy of checkpoint inhibitor therapy is driven by responses to non-self “neoantigens” (for example see a recent paper from the Schumacher http://www.ncbi.nlm.nih.gov/pubmed/25531942) . Yet specific responses against such neoantigens, in the context of a mutationally unstable tumour, might be expected to rapidly drive the emergence of escape mutations, and the outgrowth of resistant tumour clones. The profound and long lasting responses observed clinically may depend on a concomitant but transient activation of polyclonal “helper” antiself responses, bolstered by the therapeutic suppression of natural self-regulating networks.
- Tuberculosis. There is a growing appreciation that “successful” immunological control of Mycobacterium tuberculosis infection reflects a subtle balance between too strong and too weak an immune response. Indeed the “caseating granuloma” which epitomises tuberculosis is a classic example of damage to self resulting from responses to foreign antigens. The hypotheses outlined above suggest that the robust and chronic stimulation of anti-mycobacterial responses may drive a concerted antiself response which will overcome the normal homeostatic regulatory mechanisms and result in autoimmunity and cell death.
- HIV. Despite decades of research, the exact mechanism which drives AIDS pathogenesis remain unclear. In particular, the rapid turnover of T cells far exceeds the predicted number of infected T cells, and suggests an important role for large scale “bystander” killing of uninfected T cells. As in the case of tuberculosis, the persistent strong immune response to HIV antigens may activate a sustained and pathogenic antiself response, which will contribute to the gradual erosion of the T cell compartment.
The hypotheses and these illustrative examples of how they may operate were stimulated by a purely theoretical paper, a relative rarity in the immunological literature. Theoretical physics has a well-established and respected role within the physics corpus. In contrast, theoretical biology (including theoretical immunology) remains very much a poor relation (the Journal of Theoretical Biology’s most recent impact factor was a modest 2.1). It is true that in the absence of experimental data, the hypotheses presented remain speculative. Nevertheless, I hope that putting these ideas in the public domain may stimulate experiments which will support, or perhaps falsify the hypotheses. It is time our concepts of self-nonself balance were revisited.
Single cells and their stochastic perambulations
By regfbec, on 30 June 2015
I returned last week from a small but intense meeting entitled “Stochastic single-cell dynamics in immunology : experimental and theoretical approaches”. The meeting took place in the splendid 17th century Tripenhuis building in Amsterdam, the home of the Royal Netherlands Academy of Arts and Science for over two centuries. The meeting was organised by Leïla Perié, Johannes Textor and Rob de Boer (all from the University of Utrecht) and Ton Schumacher (NKI Amsterdam).
Perhaps unsurprisingly, the meeting was driven by technical and methodological advances, both experimental and computational. Dirk Busch (TUM, Munich) presented further results from his elegant single cell adoptive transfer in vivo model system, which has already revealed the striking heterogeneity in both proliferative and differentiation fate which arise from different naïve T cells or the same specificity. A nice extension of the system was the introduction of dendritic cells carrying the diphtheria toxin receptor, which could be depleted at fixed time after immunisation, thus allowing the role of persistence of antigen to be dissected. Remarkably, perhaps, the mechanistic source of the stochastic variation remains incompletely understood, and both this group and other groups are looking to in vitro models to give a more precise handle on this fundamental question.
Another highlight of technological virtuosity was, as ever, Ido Amit’s (Weizmann Institute, Israel) presentation of his experimental system which allows global transcriptomic and chromatin modification analysis to be carried out on ever smaller numbers of cells. Single cell transcriptomics of blood has revealed an increasingly complex mixture of heterogenous “subpopulations”, although the challenge of conceptualising meaningful “shapes” in such vertiginously high dimensional space remains.
A very considerable effort from many groups focused on improving the length of time, and the precision, with which cells could be imaged in vitro. The prodigious ability of T cells to race around in three dimensions provide special challenges for imaging, which Philippe Bousso (Institut Pasteur, France) tackled by forcing them to migrate up and down narrow microchannels (reminding me rather of Olympic swimmers in their allotted lanes), thus allowing very accurate quantitation of migration velocities in one dimension. Tim Schroeder (ETH, Switzerland) presented another very elegant experimental system to tackle the challenge arising from the very slow turnover kinetics of stem cells, which therefore requires very long tracking times (in the order of weeks) in order to follow cells through differentiation. Although perhaps relying less on technical virtuosity, I liked Gregoire Altan-Bonnet’s (Memorial Sloan-Kettering Cancer Centre, USA) approach of extracting information from the natural heterogeneity captured by most flow cytometry data sets, which is otherwise largely ignored by the thousands of practitioners of flow cytometry. He illustrated how a combination of this data analysis with elegant and simple mathematical modelling can be used to extract functional regulatory networks from quite “low-tech” data sets.
Technology aside, what were the conceptual questions emerging from the meeting ? Questions of lineage loomed very large. Tracing cell family trees (questions of progenitors, descendants , branch points) has long exercised immunologists and haematologists, and new technologies have certainly promised to provide more accurate and detailed answers. In the context of the very real prospect of successful clinical stem cell therapies such questions are surely important, although the nagging concern over lineage stamp collecting remains for me a niggling concern. An idea raised by several speakers was the “stemness” properties of the T memory cell. For this writer, the idea that memory cell “self renew” seemed almost tautological. And in this context, it seemed astonishing that teleomeres scarcely got a mention ! Another surprising emphasis was the re-emergence of the concept that T cells can affect neighbouring cells through cytokine secretion. This idea, which was in some way the fundamental concept underlying the prolonged era of cytokine discovery, has gradually been replaced by the concept of specific targeted secretion dominated by tight immunological junctions. But the wheel, it appears, is about to turn full circle. A particularly intriguing extension of this is the suggestion that cytotoxic T cell mediated killing is often mediated at a distance via interferon gamma secretion. Further experimental and quantifiable evidence on the importance of this process in vivo is urgently required.
I did not emerge from this meeting with the feeling of having heard any major specific breakthrough, but rather with the sensation that the meeting represented a gathering of pioneers who are exploring the possibilities of a new type of immunology. The common theme among the participants was a willingness to engage at the frontier between computational and experimental science. The rules of this new subject, how to develop it, which approaches will be fruitful, and which will turn out to be relics of the physical sciences with little relevance to the world of cell biology : all these questions remain to be answered. But it is encouraging to find a group of scientists seriously engaged in this exciting challenge (discussion throughout the meeting was lively). Many of the senior investigators told me they faced a continual battle to secure funding and to ensure high impact publication in the face of a scientific establishment which often pays lip service to interdisciplinary research, but doesn’t understand or appreciate it when it actually finds some. In this context, one can hardly avoid mention of the enormous influence of Rob de Boer and his ever growing school of talented and imaginative disciples on the field as a whole.
I finish with a plea to conference organisers. Can we introduce a rule that any talk must present one, or at absolute maximum, two key findings ? Perhaps its my limited attention span, but learning one really important new thing from a talk is about as much as I can take in or remember. And anyway, how many of us have the privileges of discovering more than one really important thing at a time?
On the spread of viruses : the enemy within.
By regfbec, on 13 April 2015
I hope all readers saw our recent paper in PLOS Computational Biology where we explored the parallels between spread of HIV within the body , which uses a combination of cell-to-cell and cell-blood-cell transmission and the spread of computer worms, some of which use a combination of local neighbourhood and remote probing to achieve maximal efficiency and persistence. Here I would like to extend the discussion of these results to a more speculative level than is generally allowed in the confines of rigorous academic peer review. I apologise that this discussion is light on quantitative analysis, contrary to the spirit of the blog. But perhaps someone can propose some quantitative way to address the questions I raise below.
The first question I would like to pose is from the perspective of HIV. What advantage, from an evolutionary perspective, is provided by dual spreading modes for the virus ? There is clearly a substantial cost to the virus involved in producing high levels of secreted virions, the vast majority of which die within 20-60 minutes without infecting another cell.
One hypothesis might be to suggest that it allows more rapid circulation of the virus and hence access to a larger reservoir of susceptible cells. Migrating cells routinely encounter hundreds, or even thousands of other T cells per hour [1,2] ) . Infected cells, however, may migrate much more slowly [3]. Furthermore activated proliferating T cells, which provide by far the most effective targets of infection, may also migrate much more slowly, and remain attached to their cognate antigen presenting cell for much of their susceptible period [3]. Nevertheless, the extent of T cell recirculation makes this hypothesis less attractive.
An alternative hypothesis to explain the evolutionary advantage of hybrid spreading might be that cell free virus is transmitted from host to host much more efficiently than cell-associated virus, since the half-life of transferred cells in an allogeneic host is likely to be in the order of minutes, and may be too short to allow the establishment of the cellular synapse which is required for cell-cell transmission. An instructive comparison is to compare HIV-1 with HTLV-1, another human retrovirus. HTLV-1 has been shown to spread almost exclusively by cell-cell transmission [4], and virus in blood is generally at undetectable levels. Although the prevalence of HTLV-1 in some endemic areas is very high, the virus has an ancient history in humans, and is accompanied by a relatively indolent pathology. The virus has therefor had plenty of time to establish itself in its host population. Certainly, there is no evidence for the remarkably rapid pandemic which has characterised the spread of HIV within the last 50 years. Comparison to other viruses would surely be instructive. I am not aware of what other viruses might use two parallel modes of spread, and would love to hear of any examples.
The second question is from the point of view of computer virus security. A key component of our analysis, which is explored in more detail in two other companion papers (http://arxiv.org/abs/1409.7291; http://arxiv.org/abs/1406.6046) is that the spread of computer malaware within a local network environment, is much more efficient and rapid than spread between one local network and another. We did not investigate the reasons for this behaviour in detail, but plausibly the major block to spread of the virus are institutional firewalls and once these have been penetrated the ability to infect another virus within the same system is much greater. A corollary of this phenomenon is that the ability to discriminate and suppress viruses or worms within an organisation may be as important, or even more important, than preventing the original security breach in the first place. Strategies for identifying and decommissioning malaware spread within a local network (“neighbourhood watch” approaches ) poses formidable challenges. But such strategies do start with the advantage that the set of potential sources and targets of infection are limited in number and known in advance. And useful solutions could be of major benefit to preventing the catastrophic results of cybercrime even if the primary infection event itself cannot always be prevented.
- Munoz MA, Biro M, Weninger W. T cell migration in intact lymph nodes in vivo. Curr Opin Cell Biol. 2014;30: 17–24. doi:10.1016/j.ceb.2014.05.002
- Gadhamsetty S, Marée AFM, Beltman JB, de Boer RJ. A general functional response of cytotoxic T lymphocyte-mediated killing of target cells. Biophys J. 2014;106: 1780–91. doi:10.1016/j.bpj.2014.01.048
- Murooka TT, Deruaz M, Marangoni F, Vrbanac VD, Seung E, von Andrian UH, et al. HIV-infected T cells are migratory vehicles for viral dissemination. Nature. Nature Publishing Group; 2012;490: 283–7. doi:10.1038/nature11398
- Pique C, Jones KS. Pathways of cell-cell transmission of HTLV-1. Front Microbiol. 2012;3: 378. doi:10.3389/fmicb.2012.00378
