 Close
Close

Heterogeneity in the PCR
By regfbec, on 13 October 2015
Unexpected heterogeneity in the PCR highlights the importance of molecular barcoding in immune repertoire studies. Benny Chain and Katharine Best Read the paper here
The polymerase chain reaction (PCR) is probably the most widely used technique in biology today. It is based on the process which defines all living organisms, namely the template driven replication of a nucleic acid chain by a polymerase enzyme. Since each piece of DNA gives rise to two daughter strands at each cycle, the amplification is exponential. This gives PCR extraordinary sensitivity (a single DNA molecule can readily be detected). However, it also means that the degree of amplification achieved is very sensitive to the efficiency of amplification, namely the proportion of strands which are correctly replicated at each step. This efficiency is well known to depend on a whole variety of experimental factors, including the sequences of both oligonucleotide primers and template DNA. Traditional approaches to quantitative PCR therefore relied on measuring the average amplification of an unknown number of molecules of a specific target sequence, in comparison to known numbers of a reference sequence. However, the advent of massively parallel sequence technologies now allows one to actually count the number of times each a particular sequence is present in a PCR amplified mixture. Indeed counting heterogeneous DNA mixtures by sequencing lies at the heart of RNA-seq transcriptional profiling, and especially at attempts to describe the adaptive immune repertoire by T cell or B cell receptor sequencing. We have therefore re-examined the heterogeneity of PCR amplification using a combination of experimental and computational analysis.
In our paper we label each template molecule of DNA with a u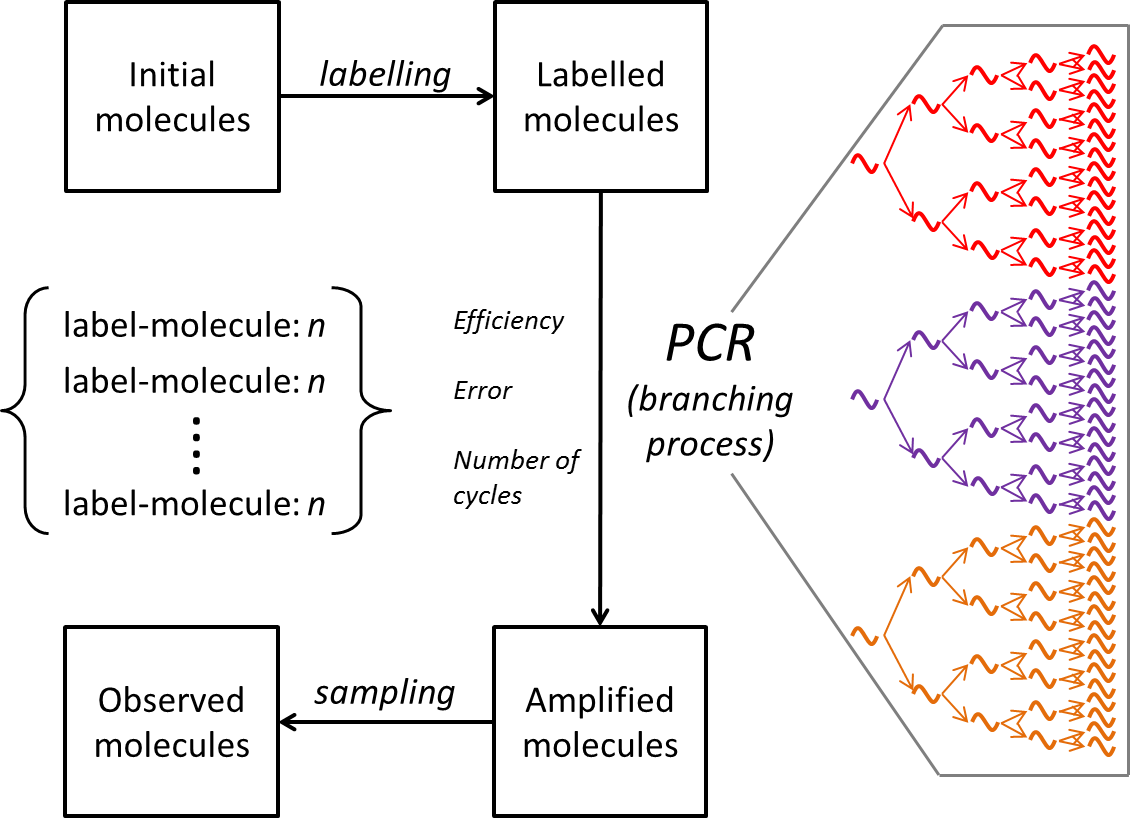 nique sequence identifier (a molecular barcode), amplify by PCR and count the number of times each DNA molecule is replicated using Illumina parallel sequencing. Remarkably, identical molecules of DNA, amplified within the same reaction tube, can give rise to numbers of progeny which can differ by orders of magnitude. PCR is an example of a branching process (see adjacent figure ). The mathematical properties of such processes have been studied quite extensively, but turn out to be complex. Indeed equations describing the processes cannot be obtained in closed form except in certain very limited specific cases. Instead, we build a “PCR” simulator (the code for which is freely available) which carries out PCR reactions in silico, tracking the fate of each molecule though a series of cycles. Using the simulator, we demonstrate that the observed heterogeneity can only be explained if different molecules of identical DNA are replicated with different efficiencies, and these differences are inherited by the progeny at ach cycle. The physical explanation of this heterogeneity remains mysterious. But these results highlight that the results of single molecule counting following PCR must be treated with caution, and highlight the importance of molecular barcoding in achieving reliable quantitative descriptions of complex molecular mixtures such as those representing the vertebrate adaptive immune repertoire.
nique sequence identifier (a molecular barcode), amplify by PCR and count the number of times each DNA molecule is replicated using Illumina parallel sequencing. Remarkably, identical molecules of DNA, amplified within the same reaction tube, can give rise to numbers of progeny which can differ by orders of magnitude. PCR is an example of a branching process (see adjacent figure ). The mathematical properties of such processes have been studied quite extensively, but turn out to be complex. Indeed equations describing the processes cannot be obtained in closed form except in certain very limited specific cases. Instead, we build a “PCR” simulator (the code for which is freely available) which carries out PCR reactions in silico, tracking the fate of each molecule though a series of cycles. Using the simulator, we demonstrate that the observed heterogeneity can only be explained if different molecules of identical DNA are replicated with different efficiencies, and these differences are inherited by the progeny at ach cycle. The physical explanation of this heterogeneity remains mysterious. But these results highlight that the results of single molecule counting following PCR must be treated with caution, and highlight the importance of molecular barcoding in achieving reliable quantitative descriptions of complex molecular mixtures such as those representing the vertebrate adaptive immune repertoire.
Benny Chain and Katharine Best
